
Leave a Message
We will call you back soon!
 Your message must be between 20-3,000 characters!
Your message must be between 20-3,000 characters!
Please check your E-mail!
SUBMIT

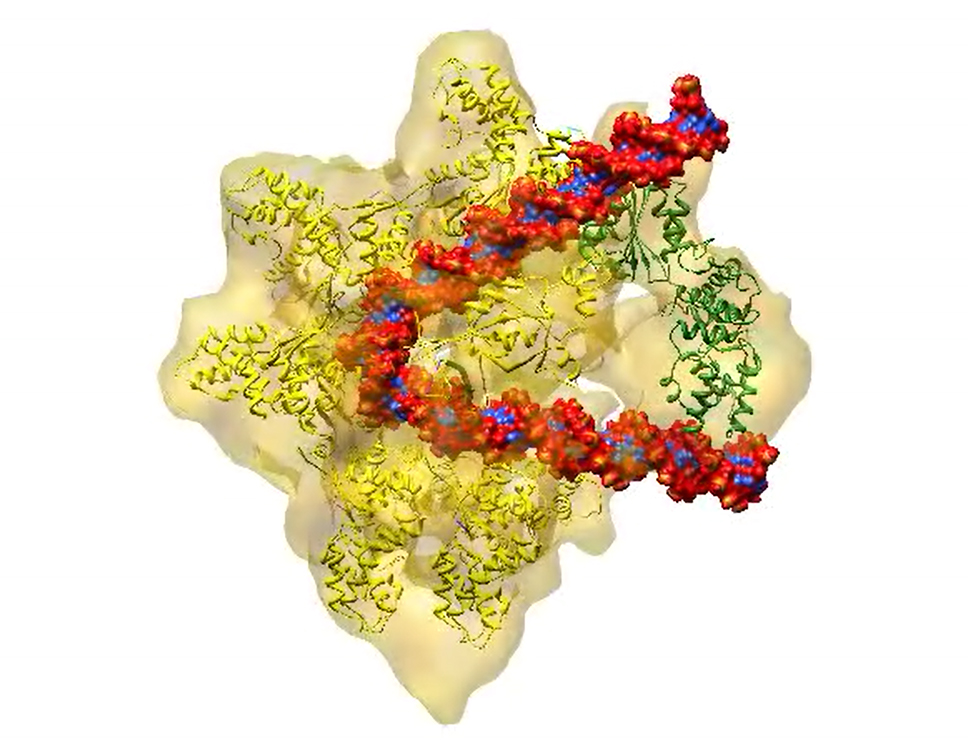
What is cryo-EM, and how does it work?
In cryo-EM, researchers rapidly freeze a cell, virus, molecular complex, or other structure so that water molecules do not have time to form crystals. This preserves the sample in its natural state. Scientists use an electron microscope to blast the frozen sample with an electron beam. This creates a two-dimensional projection of the sample on a digital detector. By creating hundreds of projections of the sample from many different angles and then taking the average of these angles, scientists generate a three-dimensional model of its structure. Recent advances in cryo-EM provide highly detailed images of proteins and other biological structures, including larger structures such as RNA-protein complexes.
Introdution of Cryogenic electron microscopy (Cryo-EM):
Over the past decade, cryogenic electron microscopy (cryo-EM) has increasingly replaced the traditional methods of sample preparation for electron microscopy. It was the pioneering work of Taylor and Glaeser and of Dubochet and colleagues that paved the way for this development, which presented a quantum leap of biological electron microscopy as it enabled to obtain images of fully hydrated specimens in a close-to-native state.
The term cryo-EM refers to various transmission electron microscopic imaging modalities when applied to samples embedded in vitreous ice. Three major branches of cryo-EM are relevant in the context of molecular structural biology: electron crystallography, single particle analysis and electron tomography.
Electron crystallography offers the advantage in determining the structure of proteins forming 2D-crystals below 4Å (e.g. as shown with water transporting membrane proteins - aquaporins). Membrane proteins are particularly promising candidates for the formations of 2D-crystals. Resolutions beyond 2 Å have been obtained. Most recently, three dimensional electron crystallography of protein microcrystals (microED) has been developed and is showing promises in solving high resolutions structures of proteins forming tiny 3D-crystals (200 nm) usually not amenable for study by x-ray crystallography.
Single particle analysis is used for isolated and purified larger assemblies of multiple subunits that are often very heterogeneous, metastable and extremely hard to crystallize (e.g. the ribosome or the 26S proteasome) at a routine resolution 3–10 Å and in best cases below 2 Å. Sample size range: 5-150 nm.
What is X-ray crystallography, and how does it work?
X-ray crystallography shoots a beam of X-rays through a tiny solid crystal made up of trillions of identical protein molecules. The crystal scatters the X-rays onto an electronic detector, similar to the way images are captured in a digital camera. A computer gauges the intensities of the scattered X-rays to assign a position to each atom in the crystallized molecule. The result is a three-dimensional digital image. This method has been used to determine more than 85 percent of known protein structures.
Some details about X-Ray Crystallography and Cryo-EM:
1. Small proteins are more suitable for x-ray crystallography since they can be difficult to discern by cryo-EM.
2. X-ray crystallography can obtain very high resolutions, although cryo-EM has had comparable results in recent years.
3. Large structures, complexes, and membrane proteins can be difficult to crystallize. Cryo-EM might therefore be an easier option.
4. EM (negative stain) offers rapid screening of samples to rule out aggregation and determine oligomerization, giving a visual overlook of how the sample behaves and what it contains.
5. Cryo-EM requires lower protein amounts than x-ray crystallography. This method might benefit samples with a yield of under 2 mg protein (<5-10 mg/ml).*
KS-V Peptide Cryo-EM Services Platform:
What is cryo-EM, and how does it work?
In cryo-EM, researchers rapidly freeze a cell, virus, molecular complex, or other structure so that water molecules do not have time to form crystals. This preserves the sample in its natural state. Scientists use an electron microscope to blast the frozen sample with an electron beam. This creates a two-dimensional projection of the sample on a digital detector. By creating hundreds of projections of the sample from many different angles and then taking the average of these angles, scientists generate a three-dimensional model of its structure. Recent advances in cryo-EM provide highly detailed images of proteins and other biological structures, including larger structures such as RNA-protein complexes.
Introdution of Cryogenic electron microscopy (Cryo-EM):
Over the past decade, cryogenic electron microscopy (cryo-EM) has increasingly replaced the traditional methods of sample preparation for electron microscopy. It was the pioneering work of Taylor and Glaeser and of Dubochet and colleagues that paved the way for this development, which presented a quantum leap of biological electron microscopy as it enabled to obtain images of fully hydrated specimens in a close-to-native state.
The term cryo-EM refers to various transmission electron microscopic imaging modalities when applied to samples embedded in vitreous ice. Three major branches of cryo-EM are relevant in the context of molecular structural biology: electron crystallography, single particle analysis and electron tomography.
Electron crystallography offers the advantage in determining the structure of proteins forming 2D-crystals below 4Å (e.g. as shown with water transporting membrane proteins - aquaporins). Membrane proteins are particularly promising candidates for the formations of 2D-crystals. Resolutions beyond 2 Å have been obtained. Most recently, three dimensional electron crystallography of protein microcrystals (microED) has been developed and is showing promises in solving high resolutions structures of proteins forming tiny 3D-crystals (200 nm) usually not amenable for study by x-ray crystallography.
Single particle analysis is used for isolated and purified larger assemblies of multiple subunits that are often very heterogeneous, metastable and extremely hard to crystallize (e.g. the ribosome or the 26S proteasome) at a routine resolution 3–10 Å and in best cases below 2 Å. Sample size range: 5-150 nm.
What is X-ray crystallography, and how does it work?
X-ray crystallography shoots a beam of X-rays through a tiny solid crystal made up of trillions of identical protein molecules. The crystal scatters the X-rays onto an electronic detector, similar to the way images are captured in a digital camera. A computer gauges the intensities of the scattered X-rays to assign a position to each atom in the crystallized molecule. The result is a three-dimensional digital image. This method has been used to determine more than 85 percent of known protein structures.
Some details about X-Ray Crystallography and Cryo-EM:
1. Small proteins are more suitable for x-ray crystallography since they can be difficult to discern by cryo-EM.
2. X-ray crystallography can obtain very high resolutions, although cryo-EM has had comparable results in recent years.
3. Large structures, complexes, and membrane proteins can be difficult to crystallize. Cryo-EM might therefore be an easier option.
4. EM (negative stain) offers rapid screening of samples to rule out aggregation and determine oligomerization, giving a visual overlook of how the sample behaves and what it contains.
5. Cryo-EM requires lower protein amounts than x-ray crystallography. This method might benefit samples with a yield of under 2 mg protein (<5-10 mg/ml).*
KS-V Peptide Cryo-EM Services Platform:
